Usage
Installation
To use cp2k_helper, first install it using pip:
(venv) $ pip install cp2k_helper
You should also be able to instal ce_expansion using the following code:
(venv) $ pip install git+https://github.com/mpourmpakis/ce_expansion.git
Finally, you can install my package from Demystifying the Chemical Ordering of Polymetallic Nanoparticles. It’s a wrapper around ce_expansion with some extra functionality.
(venv) $ git clone https://github.com/mpourmpakis/CANELa_NP.git
(venv) $ cd CANELa_NP
(venv) $ pip install -e .
Other dependencies include:
ASE
Pandas
Numpy
Matplotlib
Lxml (for web scraping)
molgif
molgif is a package developed by Michael Cowan that allows you to create gifs of molecules. It uses ase and ImageMagick. The view method with the rotate option on the NP object uses molgif to create a gif of the molecule.
Package Overviews
CANELa_NP
Using CANELa_NP to Optimize the Chemical Ordering of a AuPd Nanoparticle
Importing packages:
from CANELa_NP.Nanotools import Nanoparticle
import ase.cluster as ac
Creating a bimetallic ase atoms object:
atoms = ac.Icosahedron('Au', 5)
atoms.symbols[100:] = 'Pd'
Creating a nanoparticle object:
NP = Nanoparticle(atoms)
Visualizing the non-optimized chemical ordering:
NP.core_shell_plot()
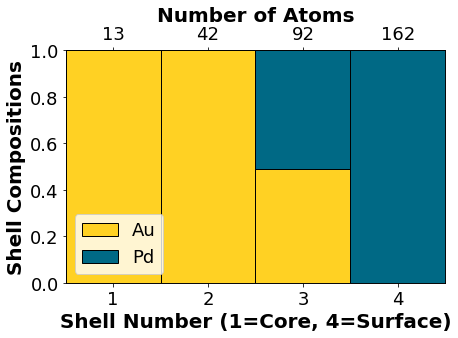
Optimizing the chemical ordering with a genetic algorithm:
NP.run_ga(max_gens=-1,max_nochange=1_000)
--------------------------------------------------
GA Sim for Au100Pd209 - none:
Min: -3.66177 eV/atom -- Gen: 02840
Form: Au100Pd209
nAtom: 309
nGens: 2840
Start: -3.44202 eV/atom
Best: -3.66177 eV/atom
Diff: -0.21974 eV/atom (6.38%)
Mute: 80.0%
Pop: 50
Time: 0:00:28
--------------------------------------------------
Saving optimized structure...
Done!
NP.core_shell_plot()
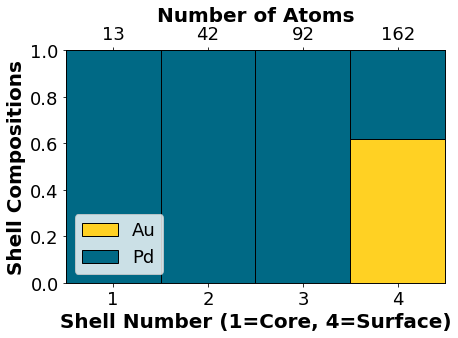
Visualizing the optimized chemical ordering (full NP):
NP.view(rotate=True,path="full_np.gif")
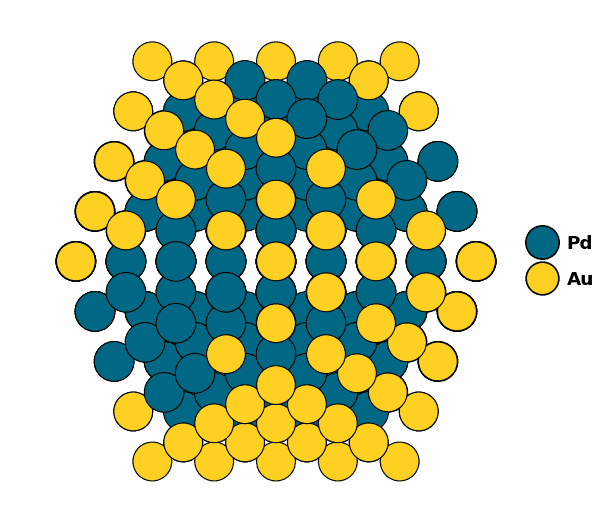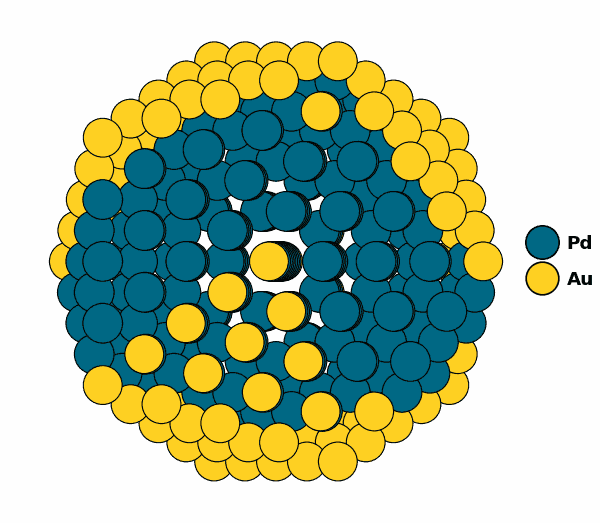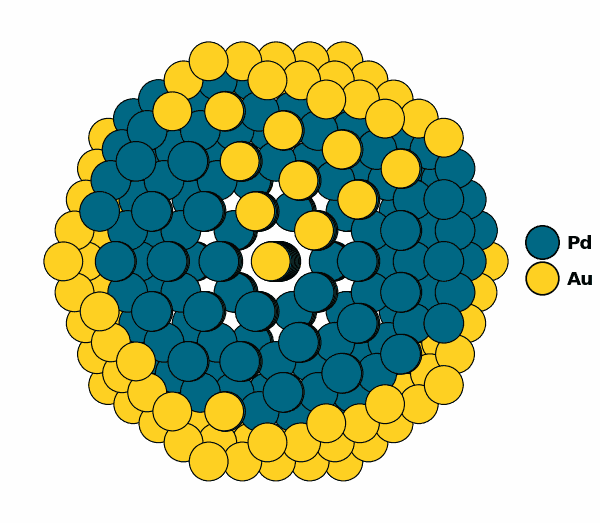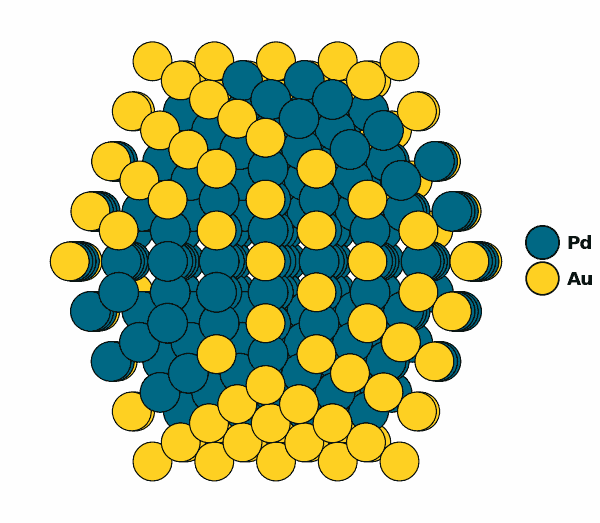
NP.view()

Visualizing the optimized chemical ordering (X-Cut NP):
NP.view(cut=True,rotate=True,path="half_np.gif")
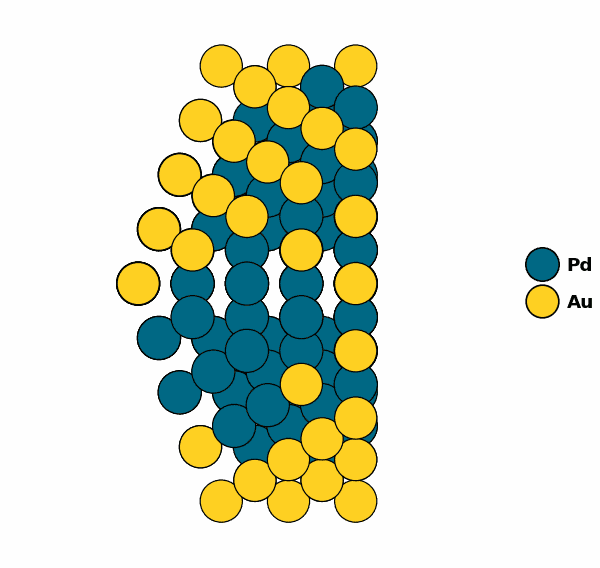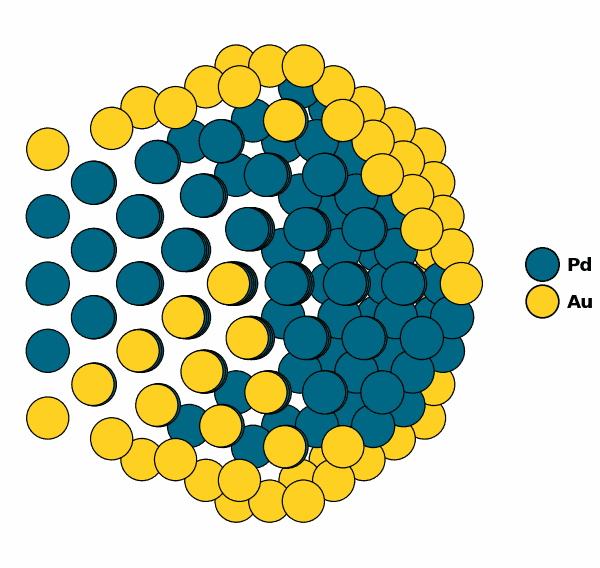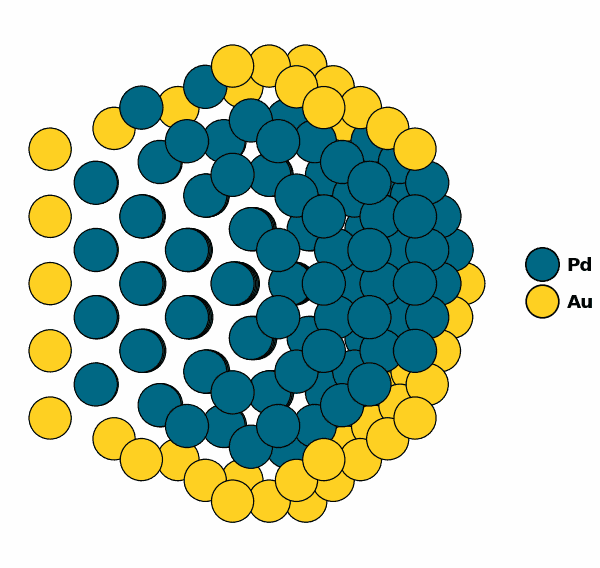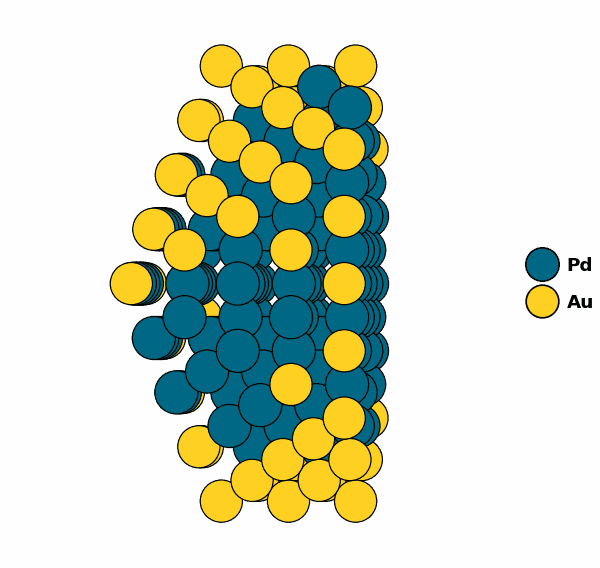
NP.view(cut=True)

Working with your own structure files
xyz_file = "Example_data/AuPdPt.xyz"
NP = Nanoparticle(xyz_file)
NP.core_shell_plot()
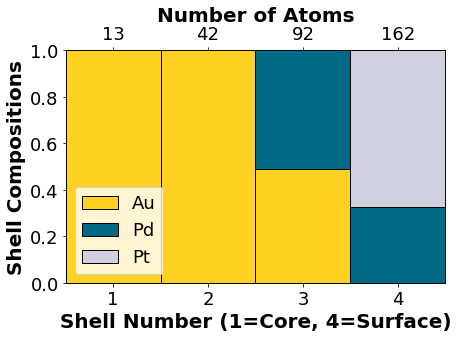
Adding your Nanoparticle Ordering to the Initial Genetic Algorithm Population
One area we have investigated is what happens when you initialize the GA with different chemical orderings. The code to do so is displayed below.
Warning
From an optimization perspective this can lead to a local minimum. It is recommended to use the fully randomized initial population as a starting point for a more robust optimization.
xyz_file = "Example_data/AuPdPt.xyz"
NP = Nanoparticle(xyz_file,spike=True)
NP.run_ga(max_gens=-1,max_nochange=1_000)
--------------------------------------------------
GA Sim for Au100Pd100Pt109 - none:
Min: -4.34346 eV/atom -- Gen: 03486
Form: Au100Pd100Pt109
nAtom: 309
nGens: 3486
Start: -4.14186 eV/atom
Best: -4.34346 eV/atom
Diff: -0.2016 eV/atom (4.87%)
Mute: 80.0%
Pop: 50
Time: 0:00:35
--------------------------------------------------
Saving optimized structure...
Done!
Calculating the Cohesive Energy of a Nanoparticle
xyz_file = "Example_data/AuPdPt.xyz"
NP = Nanoparticle(xyz_file)
NP.calc_ce()
-4.028279637969337
Calculating the Diameter of a Nanoparticle
Sometimes it is useful to know the approx. diameter of a nanoparticle. This can be done using the following code:
xyz_file = "Example_data/AuPdPt.xyz"
NP = Nanoparticle(xyz_file)
NP.get_diam() # Diameter in Angstroms
23.079965342691917
Calculating New Gamma Values
If you would like to generate gamma values for metal combinations that have not been done yet please follow the following steps from the publication Demystifying the Chemical Ordering of Multimetallic Nanoparticles by Dennis Loevlie, Brenno Ferreira, and Giannis Mpourmpakis.
Generate equally distributed NP xyz files using the script: generate_nps
Geometrically optimize these structures to find the most stable energy.
Use this script with the optimized energy values and previously generated structures to calculate the new gamma values (they will be stored in “CANELa_NP/Data/np_gammas.json”).
Above I have provided code to calculate new gamma values for metal combinations we do not currently have already. Please keep in mind that this was the code that worked for my system, I have not tested it on other systems. I have attempted to make the code general but if you are testing other systems please update the code accordingly.
Metal Type |
Used in the paper (citation below)? |
Functional |
|---|---|---|
Au |
Yes |
PBE + D3 |
Pd |
Yes |
PBE + D3 |
Pt |
Yes |
PBE + D3 |
Ag |
No |
PBE + D3 |
Cu |
No |
PBE + D3 |
Citation
If you find the code useful, please also consider the following BibTeX entry:
@article{doi:10.1021/acs.accounts.2c00646,
author = {Loevlie, Dennis Johan and Ferreira, Brenno and Mpourmpakis, Giannis},
title = {Demystifying the Chemical Ordering of Multimetallic Nanoparticles},
journal = {Accounts of Chemical Research},
volume = {0},
number = {0},
pages = {null},
year = {0},
doi = {10.1021/acs.accounts.2c00646} }
cp2k_helper
This package is a collection of functions that I use to parse and analyze CP2K output files.
Code Overview
- cp2k_helper.cp2k_helper.Summ(PATH)
This function allows you to summarize the output files from a calculation by passing in the .out file
- Parameters
PATH (str) – The path to the .out file
- cp2k_helper.cp2k_helper.checkIfDuplicates(listOfElems)
Check if given list contains any duplicates
- Parameters
listofElems (list) – List of elements to be checked for duplicates
- class cp2k_helper.cp2k_helper.output_parser(base_file_path='.', depth=inf)
Bases:
objectThis class is used to parse the output files from CP2K
- get_energies(all=True)
This function parses the OPT.out files under the given directory and outputs the energy values
- get_run_types(input_files: list) dict
This function parses the .inp files under the given directory and outputs the run type
- restart_job()
This function sets up a restart folder for the job that did not converge
- cp2k_helper.cp2k_helper.search_util(root='.', depth=inf, parse_by=None)
Recursively find all files in a directory.
Example Usage
Uses
Retreive information from the output files generated after running a calculation using cp2k.
Important Note
The class will retrieve all information under the given directory (with a max depth as an optional extra argument) and use the directory names to classify each calculation you ran. Therefore, you should not have two separate cp2k calculations with the same directory name.
Example
The output will be a dictionary of dictionaries (Containing the single point Energy calculations and Geometric optimization final energies found under the specified directory)
from cp2k_helper import output_parser
# Depth automatically set to inf
parser = output_parser(base_file_path='./cp2k')
# If all=False then only the final energies will be retrieved
Energies = parser.get_energies(all=False)
print(Energies)
Output:
{'ENERGY': defaultdict(float,
{'Folder_Name1': -1000.997638482306,
'Folder_Name2': -1000.997638482306,
'Folder_Name6': -1000.900349392778}),
'GEO_OPT': defaultdict(None,
{'Folder_Name5': -1000.900349392778,
'Folder_Name7': -1000.997638482306,
'Folder_Name3': -1000.900349392778,
'Folder_Name4': -1000.900349392778})}
Note:
The output example has fake folder names and energy values for proprietary reasons.
Command line tools
restart
cp2k_helper has a handy command line tool for restarting a calculation if it timed out. Just execute the command below in the directory that the calculation timed out and a new subdirectory will be created for the new job. You can then submit the new job to restart the calculation.
(venv) $ cp2k_helper --restart
summ
cp2k_helper can give you a quick summary of your output file. Just use the command below with your output filename:
(venv) $ cp2k_helper --summ OPT.out
energy
cp2k_helper can quickly get you the final energy values from all GEO_OPT or ENERGY DFT calculations under a specified directory. The values are converted from Ha to eV. They are saved as a csv (optionally you may name it whatever you want but the default is Energies.csv). An example of using this feature for all of the calculations under the current folder is below:
(venv) $ cp2k_helper --energy . My_Energy_Values
The above command will save a csv file to your current directory with all of the final energy values along with the type of calculation run and the folder name of each. As of now the .csv file will look similar to below (if you had 4 DFT calculations in the given directory).
Folder_Name |
Type |
Energy (eV) |
|---|---|---|
Folder_Name1 |
GEO_OPT |
-10000.34324 |
Folder_Name2 |
ENERGY |
-10000.34324 |
Folder_Name3 |
GEO_OPT |
-10000.34324 |